NucPosSimulator is simulation tool to identify
positions of nucleosomes from Next Generation Sequencing data.
NucPosSimulator utilizes a Metropolis Monte Carlo approach to determine the most probable
nucleosome position in overlapping and ambiguous DNA reads from
high through-put sequencing experiments.
In contrast to peak-calling procedures NucPosSimulator probes many
possible solutions with a Metropolis Monte-Carlo approach. Additionally, a Simulated
Annealing scheme can be applied, a heuristic optimization method, that
tries to find the optimal solution for complex positioning problems.
- Get NucPosSimulator as a stand-alone application at Download.
- Visualize your simulation results with NucPosViewer.
If you use NucPosSimulator please cite:
 Robert Schöpflin, Vladimir B. Teif,
Oliver Müller, Christin Weinberg, Karsten Rippe and Gero Wedemann:
Modeling nucleosome position distributions from experimental nucleosome positioning maps.
Bioinformatics, 29 (19), 2380-2386, 2013.
Robert Schöpflin, Vladimir B. Teif,
Oliver Müller, Christin Weinberg, Karsten Rippe and Gero Wedemann:
Modeling nucleosome position distributions from experimental nucleosome positioning maps.
Bioinformatics, 29 (19), 2380-2386, 2013.
We appreciate all suggestions, questions and bug reports. Contact details can be found here.
How it works
Data processing
Mapped paired end-nucleosome reads are imported into NucPosSimulator and transferred into frequency counts of nucleosome centers (dyad axis) in the defined locus. Hence, every read yields one nucleosome center, assumed to be located in the center of the read.
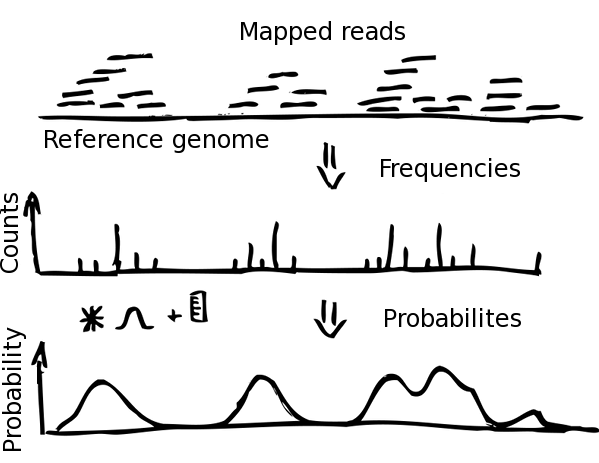
The frequency data is smoothed by a Gauss-kernel and normalized to calculate probabilities from absolute frequencies. By this, every locus position is assigned a value between 0 and 1, describing the probability that the genomic position is a center of a nucleosome. The probability function is essential for the calculation of the energy in the Metropolis Monte-Carlo simulation.
Simulation procedure
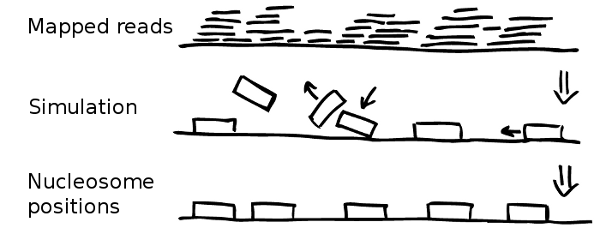
Once the experimental data is imported and processed the simulation can start. Every simulation starts with naked, i.e. nucleosome free, DNA. In the following NucPosSimulator tries to apply many times four so-called Monte-Carlo Moves to the locus:
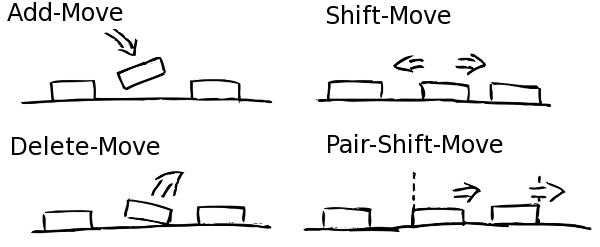
- Create a new state by applying one Monte-Carlo Move
- Compute the energy difference (∆E) between the old and the new state
- Compute the probability to accept the state: P=min(1,exp(-∆E/kT))
Accept or Reject. Step back to 1.
The moves are selected and applied randomly. Every move can be accepted or rejected according to the so called Metropolis criterion (see step 3). Therefore, the energy of the new state is computed applying an energy function. The energy function evaluates the position of all nucleosomes with respect to the underlying probability for nucleosomes according to the input data.
If the locus has a lower energy after the move, the move will be accepted immediately. If the energy of the new state is higher than before, the move is only accepted with a probability that corresponds to the energy-difference between new and old state.
If a nucleosome is removed from a more likely position, the systems energy becomes higher and it is likely that the move is rejected. If a nucleosome is shifted from a position with a low probability towards a position with a higher probability the system energy improves and the move will be accepted in any case.
Every simulation run starts with naked, i.e. nucleosome free, DNA which is populated successively. After a while the rate and with adding nucleosomes and removing is equal and the overall number of nucleosomes fluctuates around an equilibrium value.
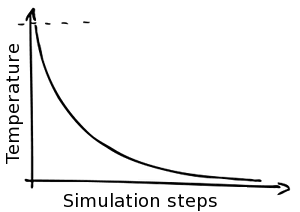
Simulated Annealing
Besides simulating at a constant temperature NucPosSimulator offers a Simulated Annealing protocol: The simulation starts at a high temperature, allowing a high dynamic and many fluctuation, even to states that are not very favorable. The temperature is cooled down stepwise, lowering the dynamics. In the end the systems freezes and only moves (e.g. movement of a nucleosome) to a more probable position are allowed. Hence, the last part of the simulations is similar to an energy optimization.